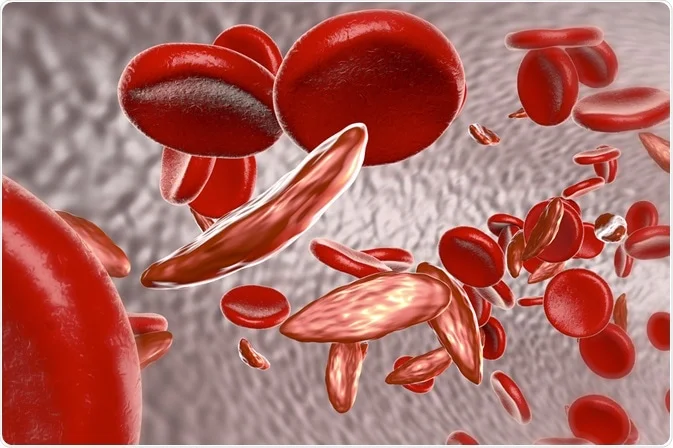
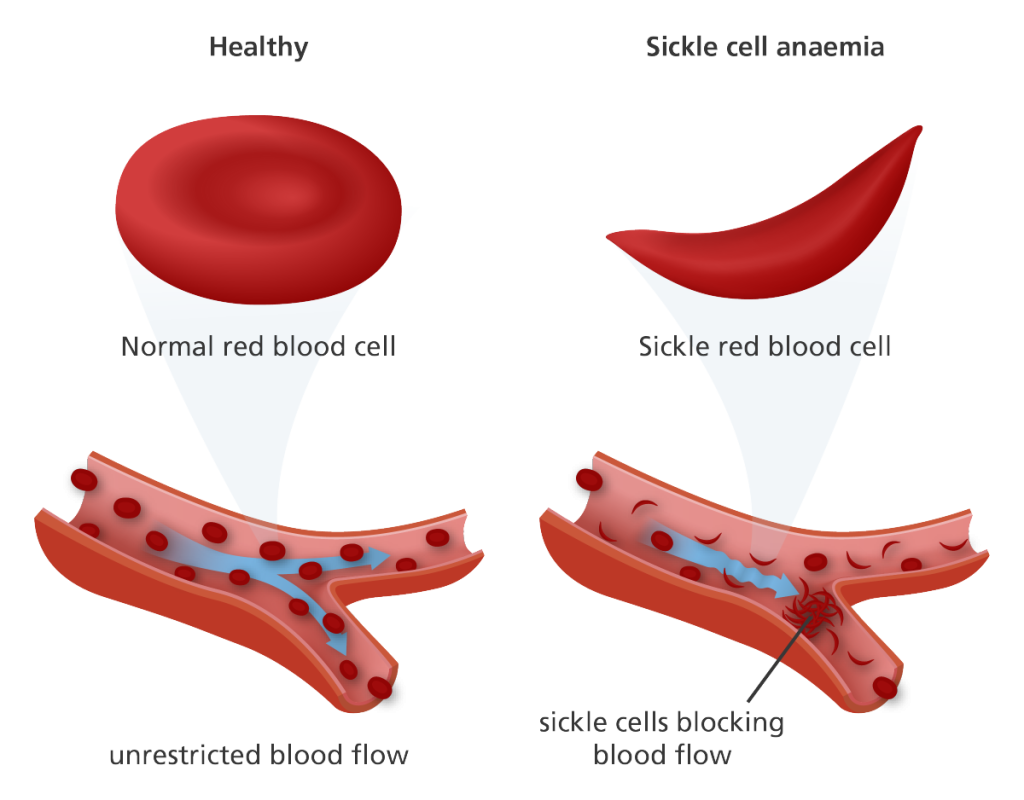
This medication is a chemotherapy drug that can help reduce the frequency of painful crises and the need for blood transfusions in some people with sickle cell disease. It works by increasing the production of fetal hemoglobin, which can prevent the sickling of red blood cells.
This medication is an amino acid supplement that has been shown to reduce the frequency of painful crises in some people with sickle cell disease.
Over-the-counter pain relievers such as acetaminophen and ibuprofen can be used to manage mild to moderate pain associated with sickle cell disease. Stronger pain medications, such as opioids, may be necessary for severe pain.
People with sickle cell disease are at an increased risk of developing infections, particularly those caused by bacteria such as Streptococcus pneumoniae. Antibiotics may be prescribed to prevent and treat these infections.
In some cases, blood transfusions may be necessary to treat complications of sickle cell disease, such as severe anemia, acute chest syndrome, and stroke.
People who receive regular blood transfusions may develop iron overload, which can damage organs such as the heart and liver. Iron chelation therapy involves taking medication to remove excess iron from the body.
As mentioned earlier, bone marrow transplant can cure sickle cell disease in some cases, although it is not suitable for everyone.
The specific medications used to treat sickle cell disease will depend on the individual’s symptoms and medical history, and should be prescribed by a healthcare provider with experience in treating the condition.